

As an expert of process design and construction in the fine chemical industry, we do our best with turn-key base system from planning to product production in order to satisfy customers.
We do directly design and bulid necessary equipments according to the customer's requirement and also directly perform process design and design and construct structures, ventilation system, electricity, and electrical instrumentation and provide customers with such high quality engineering service as after-market maintenance and service.
Bio-Diesel
Bio-Ethanol Plant
Reactor System
Distillation System
Basic &
Detail Design
Construction
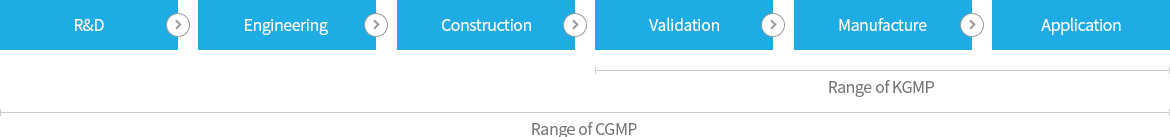
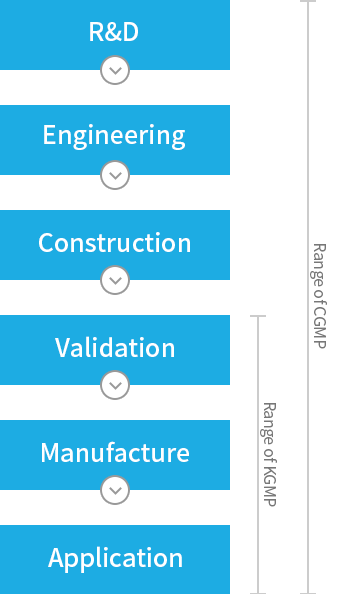
For the production of CGMP pharmaceuticals and bio products, the reproduction of efficacy, safety, and stability is essential, so such regulation agencies of advanced countries as FDA of U.S.A strictly enforce the GMP regulations to ensure them. Also, CGMP or EU-GMP corresponding to CGMP is strictly required when pharmaceutical products are imported into all advnced countries, and therefore development or securement of the technology satisfying CGMP (hereinafter CGMP technology) is vital for exporting newly developed pharmaceuticals toward global markets. CGMP means the requirements found in the legislations, regulations and administrative provisions for methods to be used in, and the facilities or controls to be used for the manufacturing, packing, and/or holding of a drug to assure that such drug meets the requirements as to efficacy, safety, and stability and has the identity and strength, and meets the quality and purity characteristics that it purports or is represented to possess.
Poor level of our domestic CGMP technology and lack of experts make a huge barrier to many domestic pharmaceutical manufacturers' efforts of exporting the products to global markets to overcome the limitaion of small size domestic market. If Korean domestic manufacturers are not able to improve the technology required for global standard certification, the export of their current products as well as newly developted biopharmaceuticals to the global market becomes impossible. Without development of CGMP technology, the R&D of new biopharmaceuticals will not be able to go beyond research lab. The KGMP, legislated by KFDA, applies now to all foods and pharmaceuticals produced and sold in Korea but its coverage is narrower and its control level also lower than CGMP. Thus, KFDA decided to enhance the domestic level of GMP up to that of CGMP till 2010 in order to secure the base of pharmaceuticals' export as well as to lift up the level of the national health and welfare level by improving the production technology of domestic pharmaceutical manufacturers. It leads to the urgent CGMP technology standardization endeavor of not only big but small and medium scale manufacturers of biopharmaceuticals.
The enhanced CGMP technology can be applied to the production and quality control processes of not only biopharmaceuticals but all other bio products comprehensively.
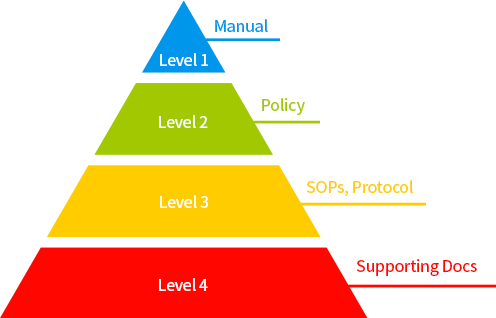
It consists of four levels.
Level 1 is
the quality manual to show important requirements and explain programs. It is a comprehensive documentation.
Level 2 is
the policy to explain the requirements for six systems and processes in a general level.
Level 3 is
the SOP and protocol to show more detailed job procedures of six systems than Level 2.
Level 4 is
other documentation not included in the above levels and it contains the real data obtained in the processes or procedures, references, and documents provided by other companies.


| Validation & EngineeringDocuments provided by equipment suppliers | Functional Specification | ||
|---|---|---|---|
| Design Specification | |||
| Design drawing | P&ID or System Diagram | Control Panel Drawing | |
| Layout | PLC Diagram(When applied) | ||
| Assembly Detail Drawing | Electricity, Control, Sequence Diagram | ||
| All other design documents | Equipment List & Engineering Calculation Data Sheet | Digital or Analog Input / Output Data | |
| Detailed Part List | Lubricant List & Schedule | ||
| Measuring Equipment List & Specification | Welding Procedure / Specification | ||
| Valve List & Specification | Cleaning / Passivation Procedure | ||
| Spatr Part List | Commissioning Part List | ||
| Sub_Vendor List | Fabrication Procedure & Time Line | ||
Submit At the stage of evaluation of design eligibility Prior to the stage of equipment fabrication Prior to purchase contract.
| The documents provided by equipment suppliers | ||
|---|---|---|
| At the stage of equipment and facility fabrication | At the stage of equipment / facility Installation and start up | At the stage of evaluation of installalation & Operation eligibility |
| FAT Protocol & Report | Installation/ Operation / Maintenance manual | Test certificate of primary components |
| Welding Quality Report (When applied) | Installation/ Operation / Maintenance manual | Material specification of primary components |
| Surface Quality Report | SAT Protocol & Report (Visual inspection, Function Test, Control Loop Test, Insulation resistance(earth) Test, Insulation Test | Calibration report of measuring instruments |
| Hydraulic Test Report (When applied) | Catalog Information of primary components | |
| Test certificate of pressure vessel(When applied) | Specification of lubricants, MSDS of coolants, thermal insulator | |
| Supplier's shipping inspection Report | Training program of Operation and maintenance of relating equipments | Results of evaluation of installation & operation eligibility point of deviation and its preventive measure report (when deviation occurs) |
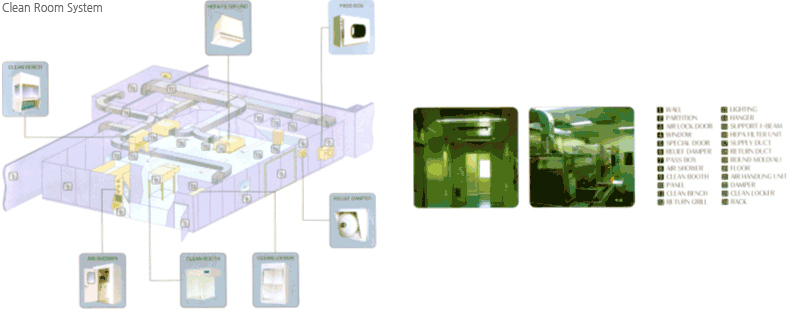
Clean Room means the system to isolate a specific space from an environment to keep dusts or minute particles within strict limits.
The specification of the clean room is defined according to maximum allowable particle diameter, and also according to the maximum allowable number of particles per unit volume. In general, the term 'class' is used for the definition - the number of particles of size 0.5mm or larger permitted per cubic foot of air.
For example, A 'Class 1000' means a space designed to never allow more than 1000 particles (0.5 microns or larger) per cubic foot of air. Generally, classrooms of class 100 or 1000 are built fot the LCD or PDP production plants while those of class 1 or 10 for semiconductor manufacturing plants.
Briefly speaking, a classroom is cleaned and maintained by forcing air flow downward from ceiling to floor in the clean room in order to drag floating dusts in air to th floor, which are removed through many holes on the floor as tiny as the size of ball point pen.
The downward air flow is circulated after particles are removed in Hepa filter installed at the clean room celiing. The clean room maintains its celanness by this circulation and air filtering. Then, what on earth is a main pollution source of a clean room? it is a human. Human body is covered with numerous tiny particles. Let alone coughing or sneezing, sweat or breath emitted by human contains millions of tiny particles. So, very strict regulation is imposed on the class room entry and exit, Which undergo several cleaning procedures and require 'smock' instead of general clothes. The clean rooms are built in R&D institutions and machinery plants producing semiconductor process machines as well as Display or semiconductor processing plants. Also, it is built in a biology lab to keep bacteria out.
BD STORAGE TANK

OFF_SITE STORAGE TANK

ACETONE STORAGE SYSTEM

E.O/P.O STORAGE SYSTEM
